Understanding the molecular characteristics and vulnerabilities of sarcomatoid/rhabdoid renal cell carcinomas through integrative histological and spatial genomics approaches
Mustafa Soytas1,4, Kate Glennon2, Minjun Kim2, Peixi Liu2, Eleonora Scarlata3, Tamiko Nishimura2, Madeleine Arseneault2, Senthilkumar Kailasam2, Fadi Brimo3, Simon Tanguay4, Yasser Riazalhosseini1,2.
1Victor Philip Dahdaleh Institute of Genomic Medicine, McGill University, Montreal, Quebec, ; 2Department of Human Genetics, McGill University, Montreal, Quebec, ; 3Division of Pathology, McGill University, Montreal, Quebec, ; 4Division of Urology, McGill University, Montreal, Quebec,
Introduction:
Genomic and immune analyzes in Sarcomatoid/rhabdoid Renal Cell Carcinoma (S/R RCC) have been limited to bulk tumor analysis and thus lack cellular resolution and spatial perspective. Herein, we use in situ Whole Transcriptome Profiling (WTP) to define molecular differences between tumor regions with and without S/R features, aiming at identifying molecular markers of S/R tumors that could lead to better diagnosis or treatments.
Methods:
All patients who underwent surgical excision of RCC at the MUHC between 2010 and 2020 were screened by a uropathologist, and histologically defined regions of S/R, ccRCC, papillary, chromophobe RCC, and benign kidney were selected to construct tissue microarrays (TMAs). Whole-exome sequencing (WES) and Compartment-Guided Spatial WTP were applied for gene and transcriptome analysis (Figure 1).
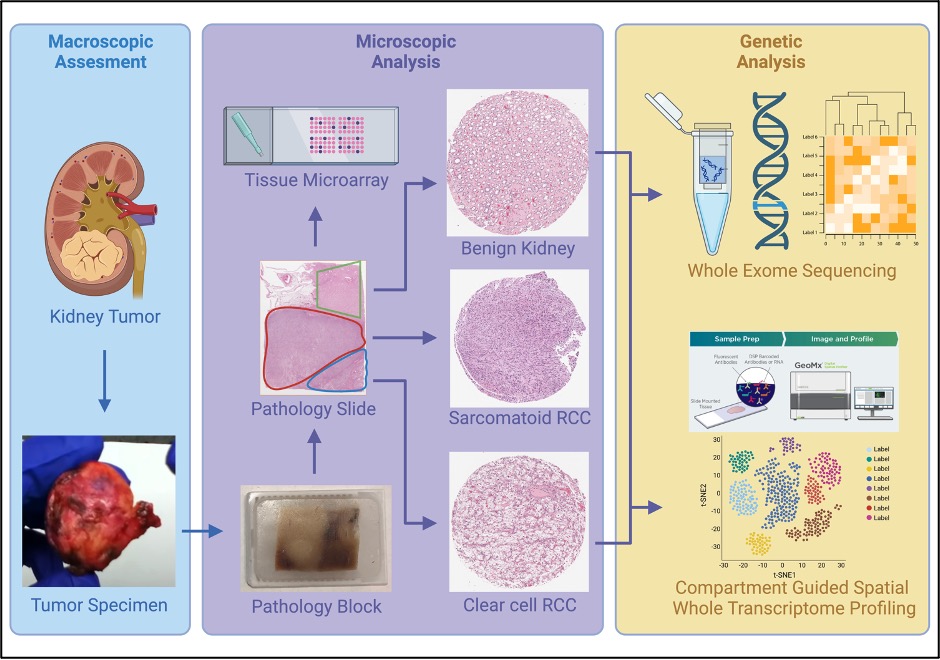
Results:
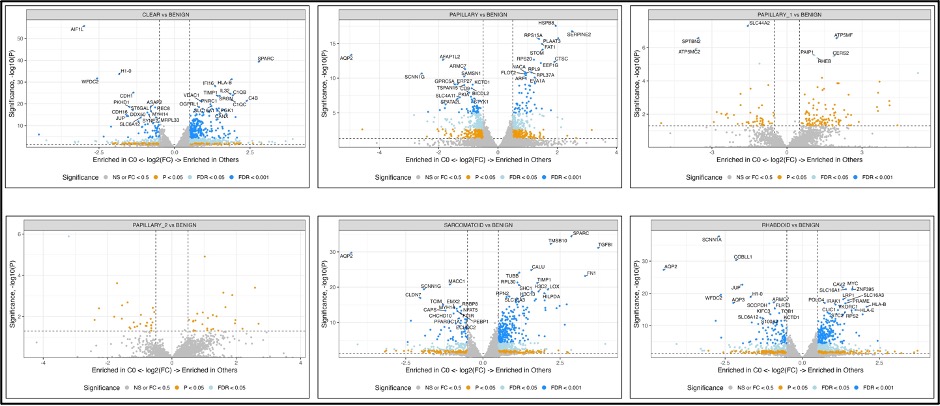
A cohort of 56 RCC patients and their TMAs, consisting of 403 cores representing patient-matched tumor areas with and without S/R features. For WES, 47 patients were used to identify copy number variations (CNVs) analysis. Four hundred cores of 55 patients were used for WTP and 5 groups of clustered with 2000 highly variable genes (HVGs) were constructed. The most variable genes of each tumor type were identified by using digital spatial transcriptome profiling (Figure 2).
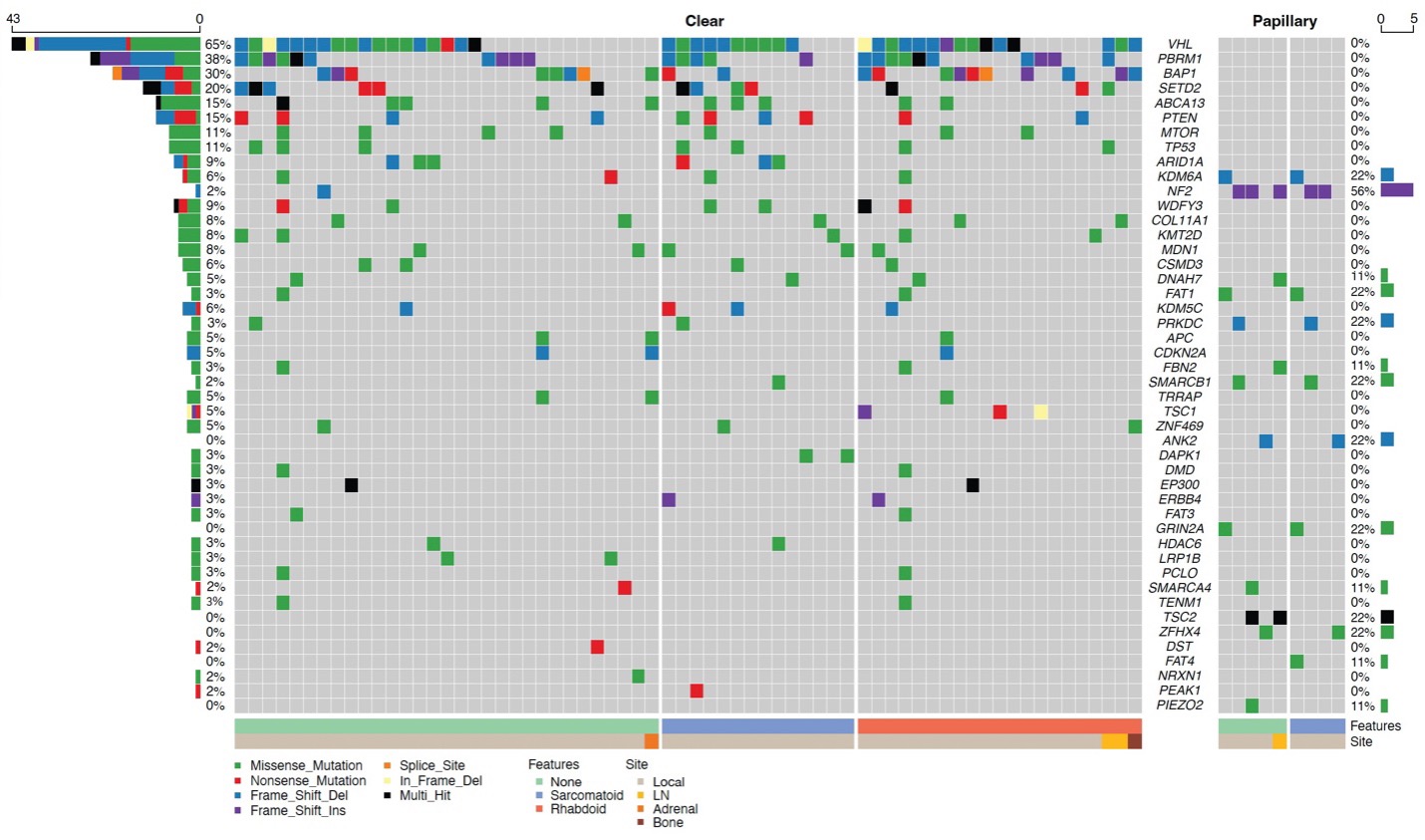
Whole-exome sequencing was used to identify mutational patterns of tumor cells using a list of specific genes of interest (Figure 3).
Conclusions:
According to current and ongoing results, WES, and compartment-guided WTP should be used to generate an unprecedented resolution to the molecular and genomic characteristics of S/R RCC tumors and tumor microenvironment.
Présentations par / Lectures by Mustafa Soytas
| Quand | Session | Titre | Salle |
|---|---|---|---|
|
ven.-08 13:20 - 14:50 |
SESSION IV | UNDERSTANDING THE MOLECULAR CHARACTERISTICS AND VULNERABILITIES OF SARCOMATOID/RHABDOID RENAL CELL CARCINOMAS THROUGH INTEGRATIVE HISTOLOGICAL AND SPATIAL GENOMICS APPROACHES | Salle de bal |